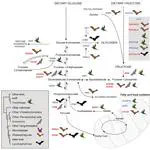
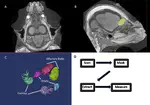
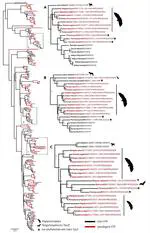
With diverse mechanical and sensory functions, the vertebrate cranium is a complex anatomical structure whose shifts between modularity and integration, especially in mechanical function, have been implicated in adaptive diversification. Yet, how mechanical and sensory systems and their functions coevolve, and how their interrelationship contributes to phenotypic disparity remain largely unexplored. To examine the modularity, integration, and evolutionary rates of sensory and mechanical structures within the head, we analyzed hard and soft tissue scans from ecologically diverse bats in the superfamily Noctilionoidea, a clade that ranges from insectivores and carnivores to frugivores and nectarivores. We identified eight regions that evolved in a coordinated fashion, thus recognizable as evolutionary modules: five associated with bite force, and three linked to olfactory, visual, and auditory systems. Interrelationships among these modules differ between Neotropical leaf-nosed bats (Family Phyllostomidae) and other noctilionoids. Consistent with the hypothesis that dietary transitions begin with changes in the capacity to detect novel food items followed by adaptations to process them, peak rates of sensory module evolution predate those of some mechanical modules. We propose the coevolution of structures influencing bite force, olfaction, vision, and hearing constituted a structural opportunity that allowed the phyllostomid ancestor to take advantage of existing ecological opportunities and contributed to the clade’s remarkable radiation.
 Photo by Stephen Rossiter
Photo by Stephen Rossiter