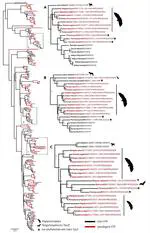
Bats possess extraordinary adaptations, including flight, echolocation, extreme longevity and unique immunity. High-quality genomes are crucial for understanding the molecular basis and evolution of these traits. Here we incorporated long-read sequencing and state-of-the-art scaffolding protocols1 to generate, to our knowledge, the first reference-quality genomes of six bat species (Rhinolophus ferrumequinum, Rousettus aegyptiacus, Phyllostomus discolor, Myotis myotis, Pipistrellus kuhlii and Molossus molossus). We integrated gene projections from our ‘Tool to infer Orthologs from Genome Alignments’ (TOGA) software with de novo and homology gene predictions as well as short- and long-read transcriptomics to generate highly complete gene annotations. To resolve the phylogenetic position of bats within Laurasiatheria, we applied several phylogenetic methods to comprehensive sets of orthologous protein-coding and noncoding regions of the genome, and identified a basal origin for bats within Scrotifera. Our genome-wide screens revealed positive selection on hearing-related genes in the ancestral branch of bats, which is indicative of laryngeal echolocation being an ancestral trait in this clade. We found selection and loss of immunity-related genes (including pro-inflammatory NF-κB regulators) and expansions of anti-viral APOBEC3 genes, which highlights molecular mechanisms that may contribute to the exceptional immunity of bats. Genomic integrations of diverse viruses provide a genomic record of historical tolerance to viral infection in bats. Finally, we found and experimentally validated bat-specific variation in microRNAs, which may regulate bat-specific gene-expression programs. Our reference-quality bat genomes provide the resources required to uncover and validate the genomic basis of adaptations of bats, and stimulate new avenues of research that are directly relevant to human health and disease1.