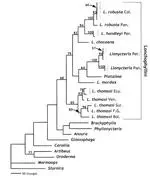
A combination of 1,140 base pairs of the mitochondrial cytochrome b gene of Platalina, Lionycteris, and several species of Lonchophylla (Chiroptera: Phyllostomidae) with 150 morphological, sex chromosome, and restriction site characters were used in an attempt to resolve relationships among the lonchophylline taxa. In addition, the monophyly of Lonchophylla was tested, particularly with respect to Platalina. The most parsimonious hypothesis of relationships using all available characters was (L. mordax ((L. chocoana (L. robusta, L. handleyi))(L. thomasi (Lionycteris, Platalina)))). Lonchophylla appears to be paraphyletic, but this arrangement is not well supported. Our analyses suggest that Platalina is not simply a large Lonchophylla, as had been suggested by previous morphological analyses. The low support values for basal relationships found in this study are probably caused by saturation in cytochrome b 3rd positions. Additionally, 2 alternative explanations are viable (if improbable): unsampled lonchophyllines are necessary to confidently resolve relationships at the base of the group, or the lack of resolution at the base of the lonchophylline phylogeny might be explained by rapid speciation following the separation from other glossophagines. Future work examining the phylogenetic relationships of lonchophylline bats should focus on describing new taxa, obtaining tissue samples from unsequenced representatives, and adding nuclear loci to this mitochondrial DNA data set.