Saturation and base composition bias explain phylogenomic conflict in Plasmodium
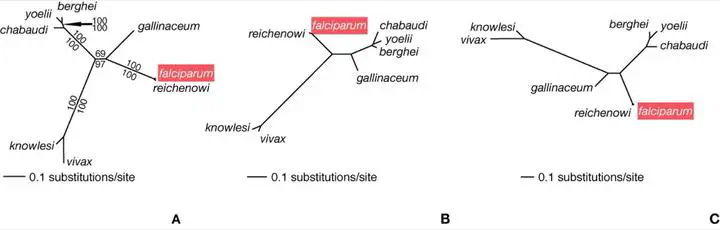 Image credit: Liliana M. Davalos
Image credit: Liliana M. Davalos
Abstract
Despite recent genome-based advances in understanding Plasmodium molecular evolution and its relationship to disease mechanisms and potential drug development, the phylogenetics of the group is currently limited to single-gene analyses. Here we develop and analyze a set of >100 putative orthologous genes derived from genome comparisons. We aimed to minimize systematic errors that arise when reconstructing the Plasmodium phylogeny with a genome-scale data set by evaluating the congruence of different genes, optimality criteria, and models of sequence evolution with previous studies encompassing fewer characters and more species. Saturation in substitutions and bias in base frequencies at third-codon positions characterized most Plasmodium genes. Molecular evolution models that partitioned rates of change by codon position were best at accounting for these sequence characteristics, as were analyses of amino acid alignments. These methods also ameliorated, but did not entirely avoid, the impact of reduced taxon sampling on phylogeny. The use of these models and expanded taxon sampling are necessary to maximize detection of multiple substitutions, overcome compositional biases, and, ultimately, resolve with confidence the phylogeny of Plasmodium.